Research
Estradiol-Mediated ER-alpha Trafficking at the Membrane
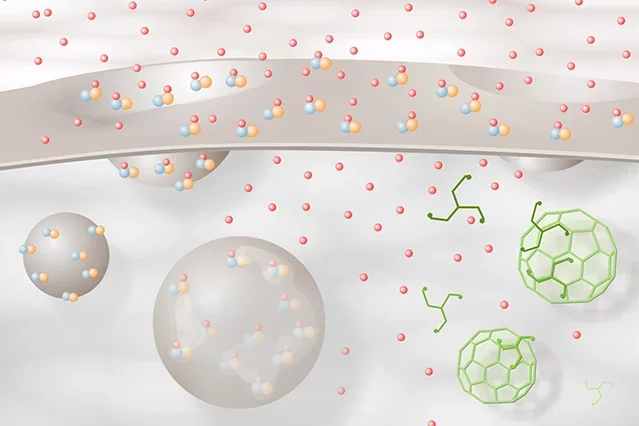
Schematic diagram illustrating estradiol-mediated ERα trafficking at the membrane. ERα-mGluR1a (blue-orange) complexes are inserted into the membrane of an exocytotic vesicle. These are associated through interactions with calveolin. Estradiol (red spheres) induces vesicle docking with the membrane and insertion of the ERα-mGluR1a complex into the membrane. Exposed to the extracellular space, estradiol binds to ERα, transactivating mGluR1a and initiating cell signaling. Estradiol-activated ERα-mGluR1a complexes are then internalized via a clathrin-mediated process and fuse with early endosomes where the estradiol is released from ERα. The ERα-mGluR1a complex can then be recycled to the membrane or degraded. Initially, there appears to be a recycling of the ERα-mGluR1a complex, but with continued stimulation, the process shifts toward degradation.
Steroid Modulation of Sexual Receptivity
Sexual receptivity is induced by the activity of estradiol (the primary estrogen in the body) through complex mechanisms that involve multiple signaling strategies in the brain. Using a rodent model, we have identified specific endogenous opioid circuits in the area of the hypothalamus that are rapidly activated by estradiol to transiently inhibit sexual receptivity, and its behavioral correlate, the lordosis reflex. In turn, progesterone, the other ovarian steroid, acts on these same circuits to reverse the inhibitory action of estradiol and facilitate lordosis. Interestingly, this transient inhibition is critical for the induction of sexual receptivity. We are studying the neurochemistry of these circuits and identifying the specific signaling pathways through which estradiol and progesterone modulate receptive behavior.
Estradiol Membrane Signaling
The GPCR/MOR internalization assay has allowed us to dissect the signaling pathways activated by estradiol. For example, in the arcuate nucleus, estradiol acts on a membrane estrogen receptor-α (ERα) to transactivate a metabotropic glutamate receptor, which phosphorylates a protein kinase-θ dependent signaling cascade responsible for lordosis circuit activation. Estradiol also regulates the levels of its cognate receptor on the membrane through modulating the trafficking of ERα to the membrane and internalization. The ERα is a classic nuclear estrogen receptor and requires a chaperone protein to be inserted into the cell membrane. Knock down of the chaperone protein caveolin-1 prevents ERα trafficking to the membrane and abrogates estradiol induced MOR activation and lordosis behavior. An intriguing and unexpected finding in these studies was the identification of an ERα splice variant, ERαΔ4 in addition to the full length ERα on the cell membrane. We are searching for the role of ERαΔ4.
Neuronal Plasticity
Estradiol also remodels the connectivity of the nuclei of the lordosis regulating circuitry. In the arcuate nucleus, estradiol induces dendritic spines, which are critical for estradiol-induced lordosis behavior. The initial spinogenesis is mediated by the stimulation of membrane estrogen receptors that deactivate (phosphorylate) an actin depolymerizing agent, cofilin. After estradiol stimulation, it takes approximately 20 hrs for the newly formed spines to become functional. We are examining the cellular and nuclear signaling pathways that underlie this estradiol induced maturation.
Estrogen Membrane Signaling in Other Systems
We have explored membrane-initiated estradiol signaling in several other systems, as well. For example, in dorsal root ganglion cells, ATP acts as a nociceptive messenger. In these cells, estradiol activates an ERα that is coupled to a metabotropic glutamate receptor that inhibits L-type voltage gated Ca2+ channels (VSCC) and attenuates the influx of calcium – thus, potentially attenuating painful signals. In a second system, the dopaminergic neurons of the substantia nigra, estradiol membrane signaling is mediated by another estrogen receptor, ERβ. Here, estradiol is neuroprotective and interacts with the IGF-1 system by activation of the phosphatidyl-inositol-3-kinase/Akt (PI3K/Akt) pathway. Estradiol acts both on neurons and through astrocytes to alter the ratio of anti-inflammatory to pro-inflammatory cytokines released may serve to protect against Parkinson’s disease.
Regulation of Neurosteroid Biosynthesis
The brain is not only a site for ovarian steroid metabolism; it is also a site of sex steroid biosynthesis. We have determined that estradiol from developing ovarian follicles stimulates the synthesis of progesterone in the hypothalamus, and this neuroprogesterone is necessary for the initiation of the LH surge. Indeed, blocking steroidogenesis in the hypothalamus of gonadally intact rats prevents the LH surge and ovulation. Males and aging females, neither of which exhibit estrogen positive feedback also lack the ability to increase progesterone synthesis in the hypothalamus. The estradiol-induced hypothalamic progesterone synthesis occurs in astrocytes. These cells express membrane ERα receptors that transactivate metabotropic glutamate receptor stimulating the release of intracellular calcium stores and augmenting progesterone synthesis.
Neuroendocrinology and Estrogen Positive Feedback
A classical question in reproductive neuroendocrinology is, “What is the mechanism of estrogen positive feedback that stimulates the luteinizing hormone (LH) surge?” This is a critical question in the field, since the LH surge triggers two central events in reproduction: ovulation and formation of the corpus luteum (which produces progesterone in the ovary). It is well-known that estrogen positive feedback is mediated by neural circuits that control the activity of gonadotropin releasing hormone (GnRH) neurons in the hypothalamus. Interestingly, in addition to estradiol, estrogen positive feedback requires progesterone – made by the brain.
"Postsynaptic" Neuroprogesterone Action
How does glial progesterone activate the neurons controlling GnRH neurons to induce the LH surge? Our working hypothesis is that kisspeptin-expressing neurons are activated by progesterone. Kisspeptin neurons have estradiol-induced progesterone receptors and potently stimulate GnRH neurons. We are currently examining the mechanism through which this glial–neuronal interaction mediates kisspeptin activation of GnRH neurons explaining estrogen positive feedback of the LH surge.
Activation of the µ-Opioid Receptor
The estradiol-induced inhibition of lordosis is mediated by µ-opioid receptors (MOR) that respond to morphine and the endogenous neuropeptide β-endorphin. In the arcuate nucleus of the hypothalamus, estradiol treatment causes synaptic release of neuropeptide Y onto POMC neurons that project to the medial preoptic nucleus and release β-endorphin to activate MOR. We study the activity of this lordosis-regulating circuit by monitoring the phenomenon of MOR receptor internalization that follows the activation of a G protein-coupled receptor (GPCR) by its endogenous ligand.